Lecture 7 Molecular Freedom and Entropy of Activation
In Lecture 6, we began thinking about the entropy of activation \(\Delta S^{\ddagger}\) in terms of changes in molecular freedom when forming the activated complex. We can develop this idea to understand systematic variations in the Arrhenius pre-exponential factor between different bimolecular reactions. We begin with a qualitative analysis before examining the molecular details more carefully.
7.1 Qualitative Analysis: Atomic and Molecular Reactions
Consider first a bimolecular reaction between two atoms. As discussed in the previous lecture, forming the activated complex reduces the number of translational degrees of freedom from 6 (3 for each atom) to 3. This loss of translational freedom corresponds to a loss of translational entropy, making \(\Delta S^{\ddagger}\) negative.
Now consider a reaction between two diatomic molecules. Each reactant molecule has 3 degrees of translational freedom and 2 degrees of rotational freedom. The activated complex has 3 translational degrees of freedom and 3 rotational degrees of freedom. Forming the activated complex now involves a decrease in the numbers of translational and rotational degrees of freedom. And we, therefore, might expect a larger, more negative, entropy of activation for this reaction than for the simpler case involving two atoms.
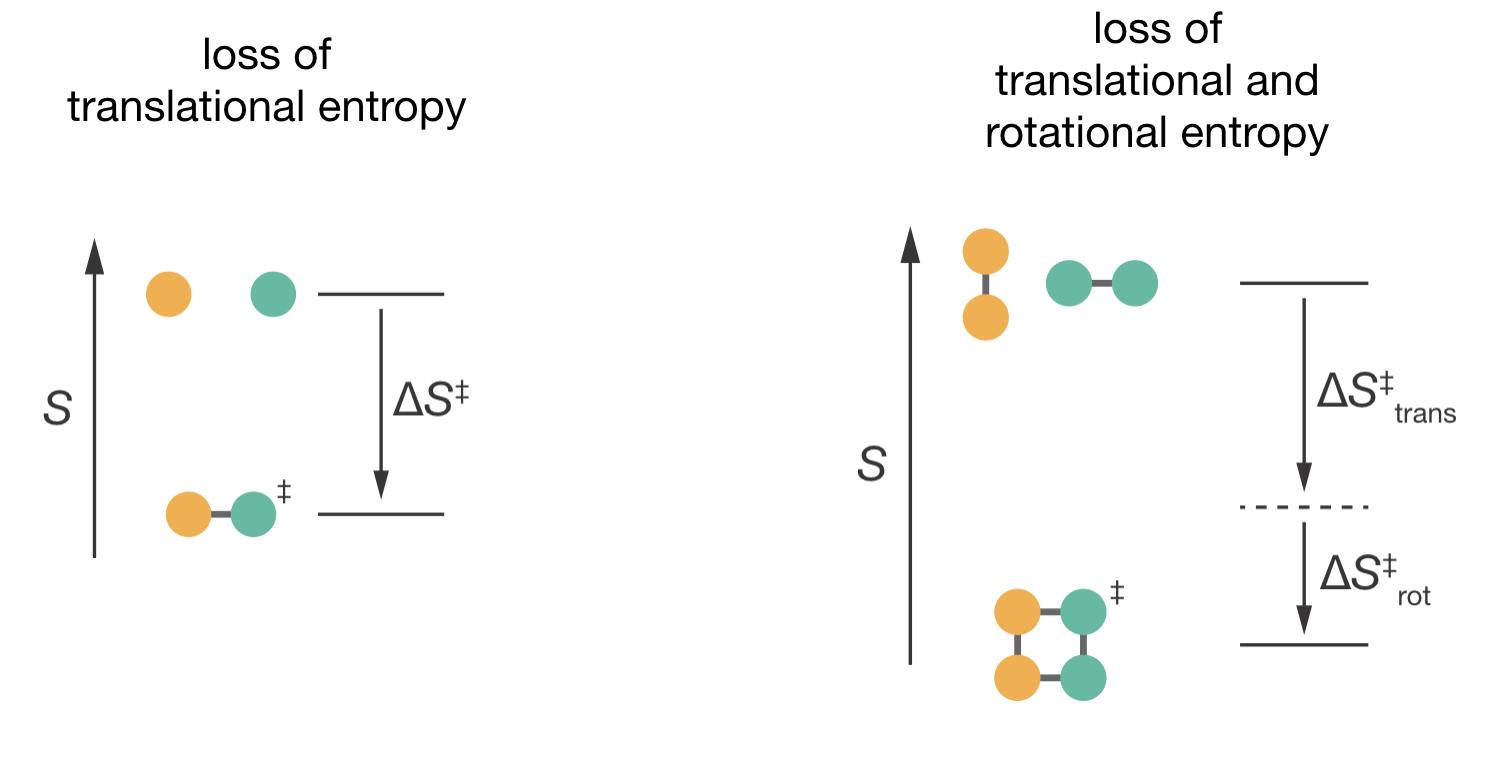
Figure 7.1: Comparing entropy changes when forming the activated complex. Left: For atomic reactions, forming the activated complex involves only loss of translational entropy. Right: For molecular reactions, forming the activated complex involves loss of both translational and rotational entropy. This additional entropic cost makes \(\Delta S^{\ddagger}\) more negative for molecular reactions, leading to smaller pre-exponential factors.
7.2 Thermodynamic Origin of the Steric Factor
In our analysis of collision theory in Lecture 5, we saw that reactions between more complex molecules typically have smaller steric factors. We interpreted this as reflecting the need for specific molecular orientations during collision. Transition state theory provides a thermodynamic perspective on this observation: in general, we can expect more complex reactant molecules to lose more molecular freedom when forming the activated complex, leading to a more negative \(\Delta S^{\ddagger}\) and consequently a smaller pre-exponential factor \(A\).
While this idea, that degree to which we lose translational and rotational degrees of freedom on forming the activated complex, is broadly correct, it is incomplete: we have not considered vibrational degrees of freedom, and we do not have a framework that can predict differences in pre-exponential factor between two molecular reactions. To proceed towards a more quantitative treatment of entropy of activation, we need a more complete accounting of all molecular degrees of freedom in the reactants and activated complex.
7.3 Molecular Degrees of Freedom
To analyse molecular freedom systematically, we begin by counting the degrees of freedom available to a molecule containing \(N\) atoms. Each atom has three degrees of freedom, corresponding to motion in three dimensions. The total number of atomic degrees of freedom is therefore \(3N\).
These atomic motions can be combined to give \(3N\) molecular degrees of freedom that describe the collective motion of the molecule.4 Molecular degrees of freedom can be classified into three types:
- Translational motion of the entire molecule through space.
- Rotation of the molecule about its principal axes.
- Vibrational motion where atoms move relative to each other.
A linear molecule has:
- 3 translational degrees of freedom.
- 2 rotational degrees of freedom.
- \(3N-5\) vibrational degrees of freedom.
A non-linear molecule has:
- 3 translational degrees of freedom.
- 3 rotational degrees of freedom.
- \(3N-6\) vibrational degrees of freedom.
7.4 A Simple Example: Atom-Atom Reaction
We can partition \(\Delta S^{\ddagger}\) into contributions from different degrees of freedom. Consider the simplest case: a reaction between two atoms, A + B, forming an activated complex C‡:
\[\begin{equation} \mathrm{A} + \mathrm{B} \longrightarrow \mathrm{C}^{\ddagger} \end{equation}\]
Let us count the degrees of freedom for the reactants and activated complex.
Reactants (two separate atoms):
Each atom can move independently in three-dimensional space, giving 3 translational degrees of freedom per atom. Since we have two atoms, the total translational degrees of freedom is 3 + 3 = 6. Atoms are point masses, so they have no rotational degrees of freedom (rotation requires at least two atoms held at a fixed distance). Similarly, single atoms cannot vibrate. Therefore, the reactants have 6 translational degrees of freedom and nothing else.
Activated complex (diatomic species):
The activated complex C\(^\ddagger\) is a diatomic species where the two atoms are connected. This single entity moves through space as a unit, giving 3 translational degrees of freedom for the centre of mass of the complex.
The complex can rotate about axes perpendicular to the bond connecting the two atoms. For a linear molecule, there are 2 rotational degrees of freedom (rotation about the bond axis itself does not change the molecule’s orientation, so only the two perpendicular axes count).
Finally, the two atoms in the complex can move relative to each other along the bond—stretching and compressing. This gives 1 vibrational degree of freedom, which corresponds to motion through the transition state along the reaction coordinate.
The total is therefore 3 + 2 + 1 = 6 degrees of freedom.
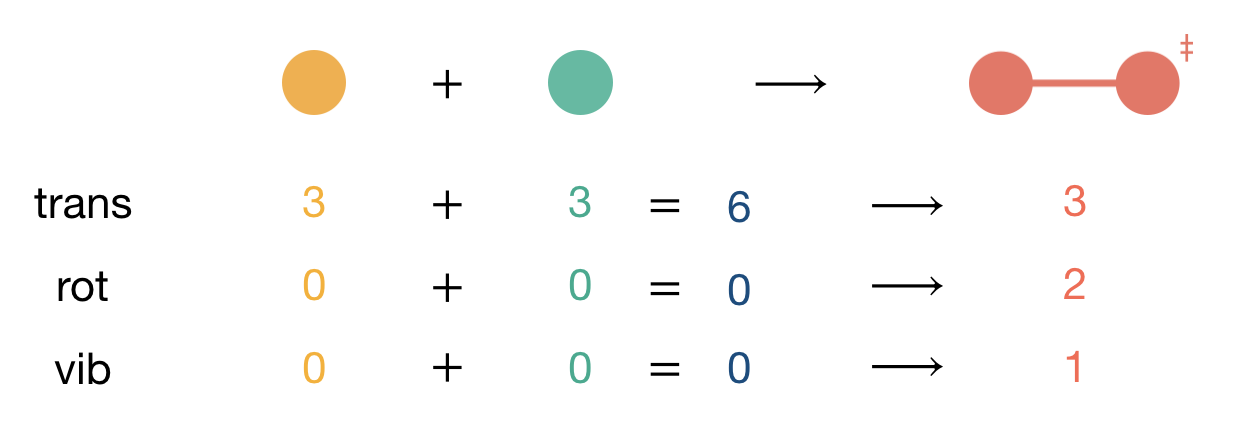
Figure 7.2: Counting degrees of freedom for the reaction A + B \(\longrightarrow\) C\(^{\ddagger}\). The total number of degrees of freedom (trans + rot + vib) is conserved: both reactants and activated complex have 6 degrees of freedom. However, the character of these degrees of freedom changes, with translational motion converted into rotational and vibrational motion.
The total number of degrees of freedom is preserved: both the reactants and the activated complex have 6 degrees of freedom. So why is \(\Delta S^{\ddagger} \neq 0\)? The answer lies in understanding that different types of molecular motion contribute very differently to entropy.
7.5 Energy Spacings and Entropy
Quantum mechanics tells us that the energy levels for molecular motion are quantised. The spacing between these levels varies significantly with the type of motion:
\[\begin{equation} \Delta \epsilon_\mathrm{trans} \ll \Delta \epsilon_\mathrm{rot} < \Delta \epsilon_\mathrm{vib} \end{equation}\]
To understand why this matters for entropy, we need the statistical mechanical foundation provided by Boltzmann’s equation:
\[\begin{equation} S = k \ln W \end{equation}\]
where \(W\) represents the number of thermally accessible microstates—the different possible arrangements available to the system at a given energy. This equation tells us that entropy increases with the number of ways energy can be distributed across the available states.
The hierarchy of energy spacings directly determines \(W\) for each type of motion. When energy levels are closely spaced, there are many thermally accessible states at typical temperatures—many different ways to distribute the available energy across the system, giving large \(W\) and hence high entropy. Conversely, when energy levels are widely spaced, only a few low-lying states can be populated, giving few ways to distribute the energy, small \(W\), and low entropy.
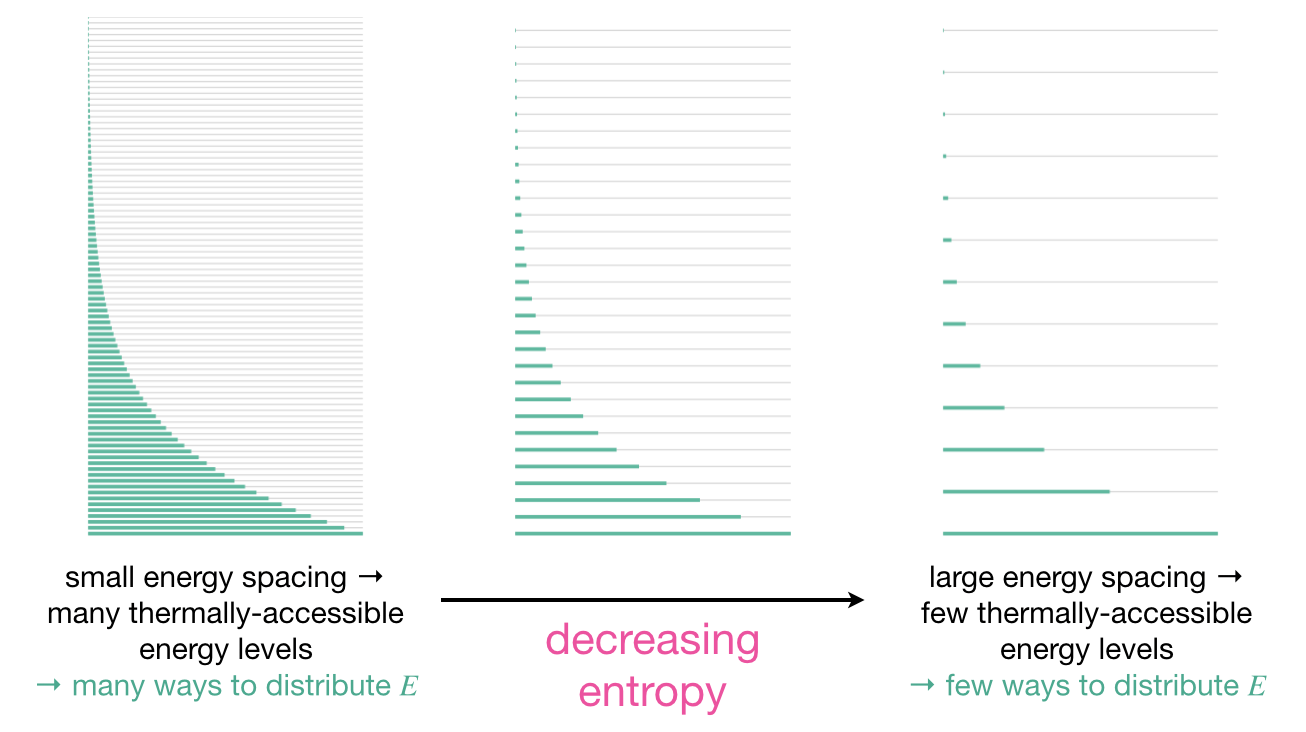
Figure 7.3: Energy level diagrams for different types of molecular motion. Small energy spacing (left) gives many thermally accessible energy levels and many ways to distribute energy, corresponding to high entropy. Large energy spacing (right) gives few thermally accessible energy levels and few ways to distribute energy, corresponding to low entropy.
For molecular motion, translational energy levels are narrowly spaced and appear almost continuous. At room temperature, an enormous number of translational states are accessible, giving very high entropy. Rotational energy levels are more widely spaced, with fewer accessible states. Vibrational energy levels are spaced more widely still, with only a few low-lying states thermally accessible at typical temperatures. This leads to the entropy hierarchy:
\[\begin{equation} S^{\ddagger}_\mathrm{trans} \gg S^{\ddagger}_\mathrm{rot} > S^{\ddagger}_\mathrm{vib} \end{equation}\]
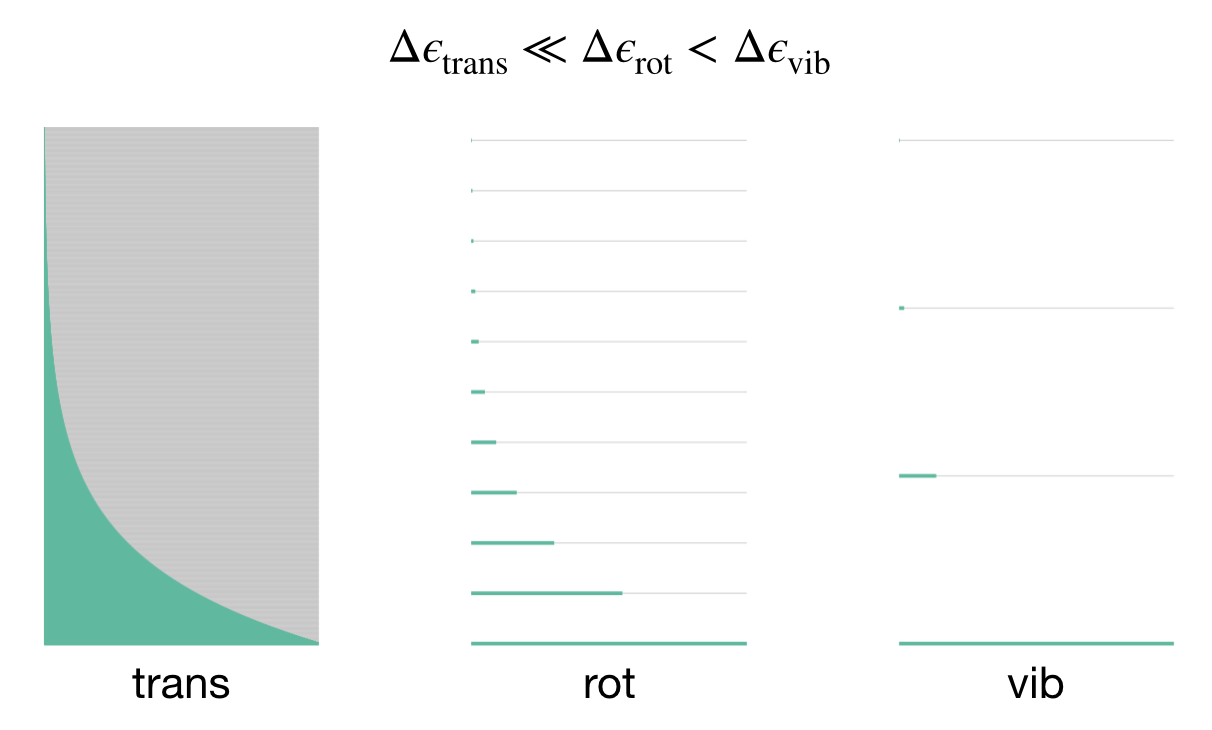
Figure 7.4: The hierarchy of energy spacings for translational, rotational, and vibrational motion. The shaded region shows the thermally accessible states at typical temperatures.
7.6 Towards Quantitative Predictions
The thermodynamic framework we have developed here provides us with a molecular perspective on the entropy of activation, \(\Delta S^{\ddagger}\). When reactants come together to form the activated complex, high-entropy molecular degrees of freedom (translation and rotation) are converted into low-entropy degrees of freedom (rotation and vibration), giving an overall negative \(\Delta S^{\ddagger}\).
To make quantitative predictions about pre-exponential factors for specific reactions, we need to estimate these entropy changes numerically. Recall from Lecture 6 that the pre-exponential factor is:
\[\begin{equation} A = \frac{k^{\ddagger}}{c^\circ} \exp\left(\frac{\Delta S^{\ddagger}}{R}\right) \end{equation}\]
In Lecture 8, we will estimate both components—the entropy change \(\Delta S^{\ddagger}\) and the rate constant \(k^{\ddagger}\)—to calculate pre-exponential factors for real reactions.
The \(3N\) molecular degrees of freedom are constructed as orthogonal linear combinations of the \(3N\) atomic degrees of freedom that are consistent with molecular symmetry. The details of this transformation from atomic to molecular degrees of freedom are part of the S2 Group Theory course↩︎